简 介
深度转录组测序能够检测数千个新的转录本。这一发现大而“隐藏”的转录组重新激活了对能够快速区分编码和非编码 RNA 的方法的需求。在这里提出了一种新的无比对方法,编码潜在评估工具( CPAT) ,可以快速识别来自大量候选转录本的编码和非编码转录本。为此,CPAT 使用了一个 Logistic 回归模型,该模型包含四个序列特征:开放阅读框大小、开放阅读框覆盖率、Fickett TESTCODE 统计量和 Hexamer 使用偏差。CPAT 软件优于其他最先进的基于比对的软件,如 Coding-Potential Calculator (sensitivity: 0.99, specificity: 0.74)和Phylo Codon Substitution Frequencies (sensitivity: 0.90, specificity:0.63)。除了高准确性外,CPAT 比 CodingPotential Calculator和Phylo Codon Substitution Frequencies 快了大约四个数量级,使其用户能够在几秒钟内处理数千个转录本。该软件接受输入序列在 FASTA 或 bed 格式数据文件。CPAT 开发了一个 web 界面,允许用户提交序列并几乎立即接收预测结果。

文件准备
这个输入文件只有一个文件可以是核酸序列文件也可以是Bed文件,例如:
>hg19_ct_UserTrack_3545_NM_001014980 range=chr1:1177826-1182102 5'pad=0 3'pad=0 strand=- repeatMasking=none
CTCGCCGCGCTGAGCCGCCTCGGGACGGAGCCATGCGGCGCTGGGCCTGG
GCCGCGGTCGTGGTCCTCCTCGGGCCGCAGCTCGTGCTCCTCGGGGGCGT
CGGGGCCCGGCGGGAGGCACAGAGGACGCAGCAGCCTGGCCAGCGCGCAG
ATCCCCCCAACGCCACCGCCAGCGCGTCCTCCCGCGAGGGGCTGCCCGAG
GCCCCCAAGCCATCCCAGGCCTCAGGACCTGAGTTCTCCGACGCCCACAT
GACATGGCTGAACTTTGTCCGGCGGCCGGACGACGGCGCCTTAAGGAAGC
GGTGCGGAAGCAGGGACAAGAAGCCGCGGGATCTCTTCGGTCCCCCAGGA
CCTCCAGGTGCAGAAGTGACCGCGGAGACTCTGCTTCACGAGTTTCAGGA
GCTGCTGAAAGAGGCCACGGAGCGCCGGTTCTCAGGGCTTCTGGACCCGC
TGCTGCCCCAGGGGGCGGGCCTGCGGCTGGTGGGCGAGGCCTTTCACTGC
CGGCTGCAGGGTCCCCGCCGGGTGGAC或者是Bed文件如下:
chr1 1370902 1378262 NM_199121 0 + 1371128 1372823 0 3 299,163,3802, 0,1799,3558,
chr1 1447522 1470067 NM_001170535 0 + 1447648 1469452 0 16 331,77,102,60,70,166,70,156,57,126,125,52,71,168,109,762, 0,3869,5168,5573,6778,7998,8405,10601,11368,11696,12130,13097,14318,15552,17080,21783,
chr1 1447522 1470067 NM_018188 0 + 1447648 1469452 0 16 331,77,246,60,70,166,70,156,57,126,125,52,71,168,109,762, 0,3869,5024,5573,6778,7998,8405,10601,11368,11696,12130,13097,14318,15552,17080,21783,在线分析
在线网址CPAT,在线使用还是非常简单,序列少可以优先选择在线操作。
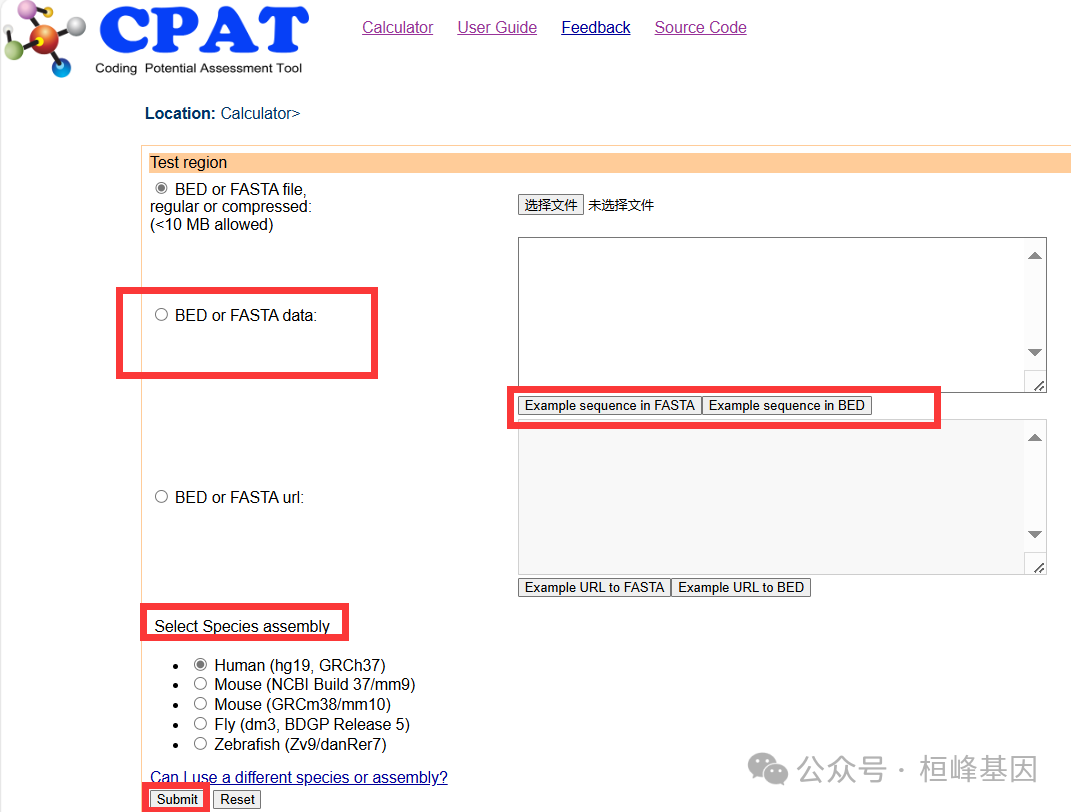
本地分析
软件包安装
安装之前要保证一些依赖的软件及包都已经准备好,python3.5 or later version,numpy,pysam,R。默认是已经安装了Anconda/miniconda3,这个需要提取配置好的哦。然后 conda 保证有python >3.5的,然后操作即可安装成功。
pip3 install CPAT实际操作
1. 参数说明
Usage:
cpat [options]Options:--version show program's version number and exit-h, --help show this help message and exit-g GENE_FILE, --gene=GENE_FILEGenomic sequnence(s) of RNA in FASTA(https://en.wikipedia.org/wiki/FASTA_format) orstandard 12-column BED(https://genome.ucsc.edu/FAQ/FAQformat.html#format1)format. It is recommended to use *short* and *unique*sequence identifiers (such as Ensembl transcript id)in FASTA and BED file. If this is a BED file,reference genome ('-r/--ref') should be specified.The input FASTA or BED file could be a regular textfile or compressed file (*.gz, *.bz2) or accessibleURL (http://, https://, ftp://). URL file cannot be acompressed file.-o OUT_FILE, --outfile=OUT_FILEThe prefix of output files.-d LOGIT_MODEL, --logitModel=LOGIT_MODELLogistic regression model. The prebuilt modelsfor Human, Mouse, Fly, Zebrafish are availablel.Run 'make_logitModel.py' to build logisticregression model for your own training datset.-x HEXAMER_DAT, --hex=HEXAMER_DATThe hexamer frequency table. Theprebuilt tables for Human, Mouse, Fly, Zebrafishare availablel. Run 'make_hexamer_tab.py' to make thistable for your own training dataset.-r REF_GENOME, --ref=REF_GENOMEReference genome sequences in FASTA format.Reference genome file will be indexed automaticallyif the index file ( *.fai) does not exist. Will beignored if FASTA file was provided to '-g/--gene'.--antisense Logical to determine whether to search for ORFsfrom the anti-sense strand. *Sense strand* (or codingstrand) is DNA strand that carries the translatablecode in the 5′ to 3′ direction. default=False (i.e.only search for ORFs from the sense strand)--start=START_CODONS Start codon (use 'T' instead of 'U') used todefine the start of open reading frame (ORF).default=ATG--stop=STOP_CODONS Stop codon (use 'T' instead of 'U') used todefine the end of open reading frame (ORF). Multiplestop codons are separated by ','. default=TAG, TAA,TGA--min-orf=MIN_ORF_LENMinimum ORF length in nucleotides.default=75--top-orf=N_TOP_ORF Number of ORF candidates reported. RNAs mayhave dozens of putative ORFs, in most cases, the realORF is ranked (by size) in the top several. It is notnecessary to calculate "Fickett score","Hexamer score" and "coding probability" for everyORF. default=5--width=LINE_WIDTH Line width of output ORFs in FASTA format.default=100--log-file=LOG_FILE Name of log file. default="CPAT_run_info.log"--best-orf=MODE Criteria to select the best ORF: "l"=length,selection according to the "ORF length";"p"=probability, selection according to the"coding probability". default="p"--verbose Logical to determine if detailed runninginformation is printed to screen.2. 构建基因组model
这个直接可以使用建好的model,在https://sourceforge.net/projects/rna-cpat/files/prebuilt_models/页面下载即可Human_Hexamer.tsv和Human_logitModel.RData
这里若是没有研究的物种,或者是基因组版本不同,那就需要自己构建model了,命令行如下:
make_hexamer_tab -c Human_coding_transcripts_CDS.fa.gz -n Human_noncoding_transcripts_RNA.fa.gz >Human_Hexamer.tsv打开Human_Hexamer.tsv文件,看一下是三列的文件:
hexamer coding noncoding
AAAAAA 0.0006471106736092786 0.001606589931772997
AAAAAC 0.00042092373222007566 0.0005113004850646316
AAAAAG 0.0008133623112408557 0.0006870944872085282
AAAAAT 0.0005917287586530271 0.0009504638599970318
AAAACA 0.0004934602747535982 0.0007256901384894673
AAAACC 0.0004003805362324795 0.0003686803641407804
AAAACG 9.064420497619743e-05 0.00010448394168197091
AAAACT 0.0004068399947646618 0.0004784022870680216
AAAAGA 0.0004286539039061299 0.0007740265969984533. 实际操作命令如下:
cpat -x Human_Hexamer.tsv --antisense -d Human_logitModel.RData --top-orf=5 -g test.fasta -o cpat_results.txt2024-07-30 03:42:59 [INFO] Running CPAT version 3.0.5...
2024-07-30 03:43:00 [INFO] Start codons used: [ATG]
2024-07-30 03:43:00 [INFO] Stop codons used: [TAG,TAA,TGA]
2024-07-30 03:43:00 [INFO] Reading Human_Hexamer.tsv
2024-07-30 03:43:00 [INFO] Checking format of "test.fasta"
2024-07-30 03:43:00 [INFO] Input gene file is in FASTA format
2024-07-30 03:43:00 [INFO] Searching for ORFs ...2024-07-30 03:43:01 [WARNING] No ORFs found for ENST000003705872024-07-30 03:43:01 [INFO] Calculate coding probability ...
2024-07-30 03:43:01 [INFO] Removing file cpat_results.txt.ORF_info.tsv
2024-07-30 03:43:01 [INFO] Select ORF with the highest coding probability ...
2024-07-30 03:43:01 [INFO] Done!结果解读
运行完成发现有6个文件出现了
Output files:*cpat_results.txt.ORF_seqs.fa: The top 5 ORF sequences (at least 75 nucleotides long) in FASTA format.*cpat_results.txt.ORF_prob.tsv: ORF information (strand, frame, start, end, size, Fickett TESTCODE score, Hexamer score) and coding probability)*cpat_results.txt.ORF_prob.best.tsv: The information of the best ORF. This file is a subset of "cpat_results.txt.ORF_prob.tsv"*cpat_results.txt.no_ORF.txt: Sequence IDs or BED entried with no ORF found.*cpat_results.txt.r: Rscript file.使用的时候就选择cpat_results.txt.ORF_prob.best.tsv文件看一下:
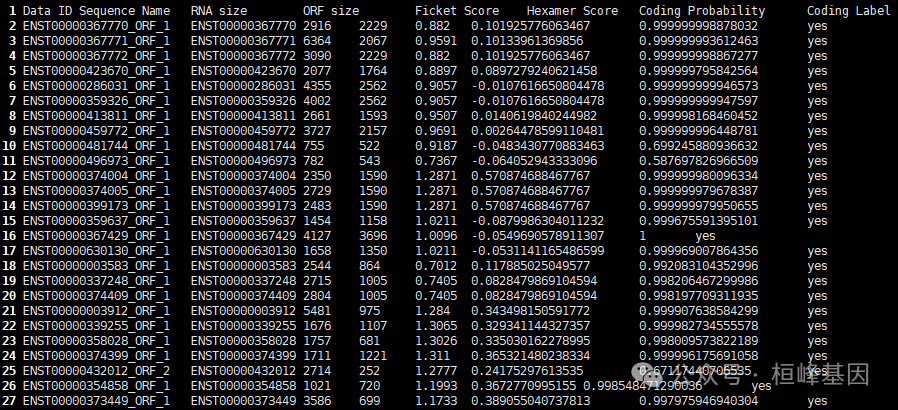
最后一列给出了转录本的蛋白编码信息,yes 代表该转录本为 protein-coding 转录本,no 代表该转录本为 noncoding 转录本。
Reference
Wang, L., Park, H. J., Dasari, S., Wang, S., Kocher, J.-P., & Li, W. (2013). CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Research, 41(6), e74.
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,
敬请期待!!
桓峰基因官网正式上线,请大家多多关注,还有很多不足之处,大家多多指正!http://www.kyohogene.com/
桓峰基因和投必得合作,文章润色优惠85折,需要文章润色的老师可以直接到网站输入领取桓峰基因专属优惠券码:KYOHOGENE,然后上传,付款时选择桓峰基因优惠券即可享受85折优惠哦!https://www.topeditsci.com/