目录
sam文件
bam文件
bed 文件
sam文件
SAM(Sequence Alignment/Map)文件是存储测序数据比对结果的一种常见格式。SAM文件通常用于存储DNA或RNA测序数据在参考基因组上的比对结果。
SAM文件由多行文本组成,每一行代表一个比对结果。SAM文件中的每一行包含了比对的序列ID、比对的标志、参考序列的名称、序列的起始位置、比对得分、序列的序列等信息。
SAM文件的格式如下所示:
头部(Header):以“@”开头的行,用来描述比对的元数据信息。
对齐记录(Alignment Records):每个对齐记录占据一行,包含了比对的详细信息。
总体来说,SAM文件提供了一个标准化的方式来存储和传输测序数据比对的结果,方便生物信息学分析和数据交换。
SAM文件的文件结构包括头部(header)和数据部分(body)。下面是SAM文件的基本结构:
1. 头部(Header):
- 头部包含一系列以“@”符号开头的元数据行,这些元数据行提供有关测序数据、参考基因组、比对算法等信息。
- 头部的元数据行可以包含信息如下:
- @HD:文件的版本和排序信息。
- @SQ:参考序列的信息,包括序列名称、长度等。
- @RG:测序样本的信息。
- @PG:比对软件的信息。
- 其他自定义的元数据行。
2. 数据部分(Body):
- 数据部分包含比对结果的详细信息,每一行代表一个比对结果。
- 每行包含多个字段,用制表符(tab)进行分隔。字段包括:
1. - QNAME:测序read的名称。
2. - FLAG:标志位,标识比对信息。
3. - RNAME:参考序列的名称。
表示read比对的那条序列的序列名称(名称与头部的@SQ相对应),如果这列是“*”,可以认为这条read没有比对上的序列,则这一行的第四,五,八,九 列是“0”,第六,七列与该列是相同的表示方法。
4.- POS:比对的起始位置。
表示read比对到RNAME这条序列的最左边的位置,如果该read能够完全比对到这条序列(CIGAR string为M)则这个位置是read的第一个碱基比对的位置,如果该read的反向互补序列比对到这条序列,则这个位置是read的反向互补序列的第一个碱基比对的位置,所以无论该read是正向比对到该序列,或是其反向互补序列比对到该序列,比对结果均是最左端的比对位置。
5. - MAPQ:比对质量得分。
表示为mapping的质量值,mapping Quality, It equals -10log10Pr{mapping position is wrong}, rounded to the nearest integer, A value 255 indicates that the mapping quality is not available. 该值的计算方法是mapping的错误率的-10log10值,之后四舍五入得到的整数,如果值为255表示mapping值是不可用的,如果是unmapped read则MAPQ为0,一般在使用bwa mem或bwa aln(bwa 0.7.12-r1039版本)生成的sam文件,第五列为60表示mapping率最高,一般结果是这一列的数值是从0到60,且0和60这两个数字出现次数最多。
6. - CIGAR:比对的CIGAR字符串,描述read和参考序列的比对关系。
CIGAR string,可以理解为reads mapping到第三列序列的mapping状态,
对于mapping状态可分为以下几类:
M:alignment match (can be a sequence match or mismatch)
表示read可mapping到第三列的序列上,则read的碱基序列与第三列的序列碱基相同,表示正常的mapping结果,M表示完全匹配,但是无论reads与序列的正确匹配或是错误匹配该位置都显示为M
I:insertion to the reference
表示read的碱基序列相对于第三列的RNAME序列,有碱基的插入
D:deletion from the reference
表示read的碱基序列相对于第三列的RNAME序列,有碱基的删除
N:skipped region from the reference
表示可变剪接位置
P:padding (silent deletion from padded reference)
S:soft clipping (clipped sequences present in SEQ)
H:hard clipping (clipped sequences NOT present in SEQ)
clipped均表示一条read的序列被分开,之所以被分开,是因为read的一部分序列能匹配到第三列的RNAME序列上,而被分开的那部分不能匹配到RNAME序列上。
"="表示正确匹配到序列上
"X"表示错误匹配到序列上
而H只出现在一条read的前端或末端,但不会出现在中间,S一般会和H成对出现,当有H出现时,一定会有一个与之对应的S出现
例如:
162M89S
162H89M
149M102S
149H102M
40S211M
20M1D20M211H
S可以单独出现,而H必须有与之对应的S出现时才可能出现,不可在相同第一列的情况下单独出现
N:如果是mRNA-to-genome,N出现的位置代表内含子,其它比对形式出现N时则没有具体解释
M/I/S/=/X:这些数值的加和等于第10列SEQ的长度。
7.MRNM
该read的mate read比对上的参考序列的名字,该名字出现在Header section的@SQ行的SN标识中,
-
如果和该read所在行的第三列“RNAME”一样,则用“=”表示,说明这对read比对到了同一条参考序列上;
-
如果mate read没有比对上,第七列则用“*”表示;
-
如果这对read没有比对到同一条参考序列,那么这一列则是mate read所在行第三列的“RNAME”。
8.MPOS
该read的mate read比对到的参考序列“RNAME”最左侧的位置坐标,也是mate read CIGAR中第一个比对标识“M”对应的最左侧碱基在参考序列的位置,未比对上的read在参考序列中没有坐标,此列标识为“0”。
9.ISIZE
表示pair read完全匹配到同一条参考序列时,两个read之间的长度,可简单理解为测序文库的长度。这个定义有两种情况(虚线表示未比对上的序列,即soft-clipped bases):
-
第一种情况如图4左所示,Read1和Read2比对到同一条参考序列,此时ISIZE即为Read2的最右侧比对坐标减去Read1最左侧比对坐标;
-
第二种情况如图4右所示,由于至今没有明确的定义和共识,因此ISIZE可以是TLEN#1,也可以是TLEN#2,视情况而定。

图片来自Sam/Bam文件格式详解
10.SEQ
存储的序列,没有存储,此列则用“*”标识。该序列的长度一定等于CIGAR标识中“M”,“I”,“S”,“=”,“X”标识的碱基长度之和。
11.QUAL
序列的每个碱基对应一个碱基质量字符,每个碱基质量字符对应的ASCII码值减去33(Sanger Phred-33 质量值体系),即为该碱基的测序质量得分(Phred Quality Score)。不同Phred Quality Score代表不同的碱基测序错误率,如Phred Quality Score值为20和30分别表示碱基测序错误率为1%和0.1%。
SAM文件是一种文本文件格式,易于人类阅读和编辑。在生物信息学中,SAM文件通常会被进一步转换成二进制格式的BAM文件,以便于存储和处理大规模的测序数据。
bam文件
BAM(Binary Alignment/Map)文件是SAM文件的二进制版本,用于存储测序数据比对结果。相对于文本格式的SAM文件,BAM文件以二进制形式存储,具有更高的存储效率和读写速度。
BAM文件格式基本上与SAM文件格式相同,但是使用了二进制编码,使得文件大小更小,并且可以更快地进行数据读写操作。BAM文件还包含了索引(index)信息,可以方便地进行随机访问和查询。
BAM文件由两部分组成:
BAM头部(BAM Header):类似于SAM文件的头部,用来描述比对的元数据信息。
BAM记录(BAM Records):每个BAM记录包含了与SAM记录相同的比对详细信息,但以二进制格式编码。
BAM文件常用于存储大规模的测序数据比对结果,例如全基因组测序、转录组测序等。它是生物信息学研究中常用的标准格式之一,方便数据的处理和共享。
BAM 文件的文件结构主要包括以下几个部分:
-
文件头(Header):BAM 文件的头部包含了一些元信息,如文件版本、参考基因组信息、测序仪器信息等。文件头通常以固定长度的MAGIC字符串开头,用于标识该文件是一个BAM文件。
-
对齐记录(Alignment records):BAM 文件的主要内容是对齐记录,每个对齐记录记录了一条测序数据的对齐信息,包括测序序列、测序质量、对齐位置等。对齐记录由多个字段组成,如QNAME(读取的名称)、FLAG(对齐标志)、RNAME(对齐到的染色体名称)、POS(对齐到的起始位置)、CIGAR(对齐方式)、SEQ(测序序列)、QUAL(测序质量)等。
-
索引表(Index):BAM 文件通常包含一个索引表,用于快速定位某个特定位置的对齐记录,加快数据检索速度。
文件头部信息通常以固定格式存储,其中包含了一系列的标签-值对(tag-value pairs),用于描述和标识测序数据的相关属性。
具体来说,BAM文件的文件头部信息通常包括以下内容:
- 申明:文件开始处包含"BGZF"四个字节的魔数标识,表示文件采用BGZF压缩格式。
- 全局头部(Header):包含了关于整个BAM文件的元信息,如文件版本号、排序顺序、参考基因组信息等。
- 参考序列(Reference Sequences):描述了BAM文件中所用到的参考基因组序列的信息,包括序列名、长度等。
- 读组(Read Groups):描述了测序数据的读组信息,如测序样本ID、测序平台、测序文库信息等。
- 标题(Comments):可以包含一些注释性的信息。
header内容不多,也不会太复杂,每一行都用‘@’ 符号开头,里面主要包含了版本信息,序列比对的参考序列信息,如果是标准工具(bwa,bowtie,picard)生成的BAM,一般还会包含生成该份文件的参数信息,@PG标签开头的那些。这里需要重点提一下的是header中的@RG也就是Read group信息,这是在做后续数据分析时专门用于区分不同样本的重要信息。它的重要性还体现在,如果原来样本的测序深度比较深,一般会按照不同的lane分开比对,最后再合并在一起,那么这个时候你会在这个BAM文件中看到有多个RG,里面记录了不同的lane,甚至测序文库的信息,唯一不变的一定是SM的sample信息,这样合并后才能正确处理。
其中,Alignment records部分包含了每条reads的比对信息,主要包括以下字段:
- QNAME:reads的唯一标识符。
- FLAG:比对标记,包括reads的比对状态和方向等信息。
- RNAME:比对到的参考基因组的染色体名称。
- POS:reads的起始位置在参考基因组上的坐标。
- MAPQ:比对质量得分,表示reads比对的可靠性。
- CIGAR:用于描述reads与参考基因组的比对关系。
- RNEXT:下一个比对的参考序列名称。
- PNEXT:下一个比对的起始位置。
- TLEN:模板长度,表示reads与其配对reads之间的距离。
Flag信息
这是一个非常特别并且重要的数字,也是一个容易被忽视的数字,这可能和许多生信工程师也并不完全理解这个值有关。许多同学在第一次看到其官方文档中的描述之后依然会觉得十分困惑,但它里面实际上记录了许多有关read比对情况的信息。想要读懂它的一个关键点是我们不能够将其视为一个数字,而是必须将其转换为一串由0和1组成的二进制码,这一串二进制数中的每一个位(注意是“位”,bit的意思)都代表了一个特定信息,它一共有12位(以前只有8位),所以一般会用一个16位的整数来代表,这个整数的值就是12个0和1的组合计算得来的,因此它的数值范围是0~2048(2的12次方,计算机科学的同学对这种计算应该不陌生)。
CIGAR
CIGAR是Compact Idiosyncratic Gapped Alignment Report的首字母缩写,称为“雪茄”字符串。它用数字和几个字符的组合形象记录了read比对到参考序列上的细节情况,读起来要比FLAG直观友好许多,只是记录的是不同的信息。比如,一条150bp长的read比对到基因组之后,假如看到它的CIGAR字符串为:33S117M,其意思是说在比对的时候这条read开头的33bp在被跳过了(S),紧接其后的117bp则比对上了参考序列(M)。这里的S代表软跳过(Soft clip),M代表匹配(Match)。CIGAR的标记字符有“MIDNSHP=XB”这10个,分别代表read比对时的不同情况:
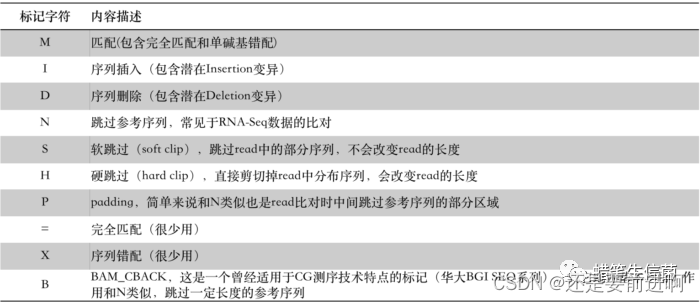
图片来自从零开始完整学习全基因组测序(WGS)数据分析之理解并操作BAM文件
除了最后‘=XB’非常少见之外,其它的标记符通常都会在实际的BAM文件中碰到。另外,对于M还是再强调一次,CIGAR中的M,不能觉得它代表的是匹配就以为是百分百没有任何miss-match,这是不对的,多态性碱基或者单碱基错配也是用M标记!
BAM文件相对于SAM文件有几个明显的优点:
存储效率:BAM文件采用二进制格式存储数据,相比文本格式的SAM文件,文件大小更小,节省存储空间。
读写速度:由于是二进制格式,BAM文件的读写速度比SAM文件更快,特别是在处理大规模测序数据时能够显著提高效率。
索引支持:BAM文件包含了索引信息,可以快速定位到特定区域或记录,方便进行随机访问和查询。
数据安全性:由于是二进制格式,BAM文件对数据的完整性和安全性有更好的保障,减少了数据损坏或篡改的风险。
格式标准化:作为生物信息学领域的标准格式之一,BAM文件具有一定的格式标准,方便数据的交换、共享和统一处理。
总的来说,BAM文件相比SAM文件在存储效率、读写速度、索引支持等方面具有明显优势,特别适用于处理大规模的测序数据以及需要高效率访问和处理数据的情况。因此,在生物信息学研究中,BAM文件被广泛应用于测序数据的存储、处理和分析。
bed 文件
BED 文件由多行组成,每行代表一个基因组区域或特征的记录。每一行包含了一系列字段,对应于区域的起始位置、结束位置以及其他属性信息。以下是 BED 格式中列的含义:
chrom:区域所在的染色体或者contig名称或编号。
start:区域的起始位置,表示在染色体上的第一个碱基位置。
end:区域的结束位置,表示在染色体上的最后一个碱基位置。
name:区域的名称或标识符。
score:区域的得分或质量值,可以表示各种数值信息,如测序深度、信号强度等。
strand:区域的方向,表示正链(+)、负链(-)或未确定链(.)。
BED 文件常用于描述基因组上的基因、转录本、外显子、修饰位点等区域,并可以携带与这些区域相关的数值数据。它在基因组注释、染色质亚结构分析和可视化等领域具有广泛的应用。
![.[[MyFile@waifu.club]].svh勒索病毒数据库恢复方案](https://img-blog.csdnimg.cn/direct/80056dcc64a540e296e96c837bd9385b.jpeg)







![[Maven]IDEA报错-xxx is referencing itself](https://img-blog.csdnimg.cn/direct/1c6ce326555b4c82888cdb6bab56914d.png)