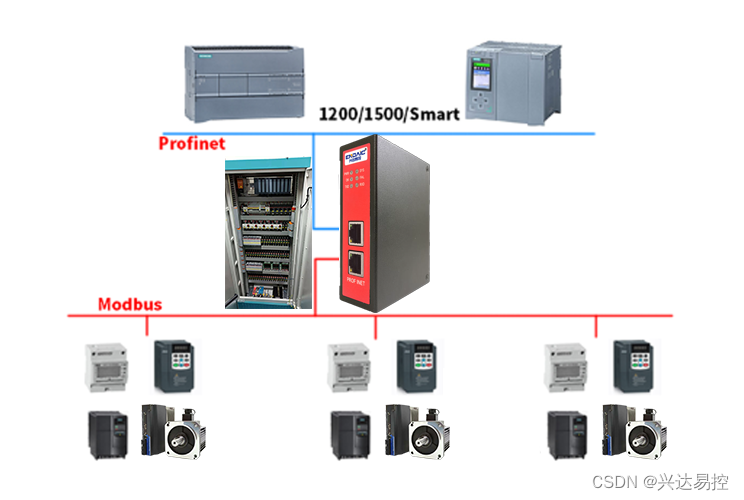
摘要:宏基因组学利用新一代高通量测序技术,以特定环境下病原体基因组为研究对象,在分析病原体多样性、种群结构、进化关系的基础上,进一步探究病原体的群体功能活性、相互作用及其与环境之间的关系,发掘潜在的生物学意义。目前,绝大部分的宏基因组学研究都集中在临床价值评价,宏基因组检测临床应用前分析性能确认的研究相对空白,北京协和医院检验科研究团队结合多年病原宏基因组检测的经验和国内外相关研究成果,就病原宏基因组项目医院本地化开展前的性能确认工作,从临床预期用途、方法学建立、性能确认、标准操作作业书4个方面提出了具体的实施方案,具体包含:标本类型和病原体范围、生物信息流程建立、生物信息分析参考盘制备和评估、湿实验流程的建立、背景核酸数据模型的建立、参考盘制备和全流程的性能确认等。
一,协和方案
病原宏基因组测序(metagenome next- generation sequencing,mNGS),基于宏基因组学和高通量测序技术,可对各种临床标本中所有的病原体(包含细菌、真菌、病毒、寄生虫、支原体/衣原体等)进行无差别检测。mNGS适用于各种病原体的鉴定,特别是未知新发病原体,如新型冠状病毒等,故在新发突发、复杂及混合感染的病原体实验室诊断中,有望提供重要的病原学诊断依据。由于mNGS仍然是一项较为前沿的技术,绝大部分相关研究多聚焦于其临床应用价值评价,分析性能确认领域相对空白。鉴于mNGS测序的湿实验和干实验过程涉及的试剂、数据库、软件等暂无标准化要求,不同实验室病原宏基因组检测流程采取的方案存在一定差异。因此,在实验室开展临床检测前,建议遵照美国临床实验室改进法案修正案(Clinical Laboratory Improvement Amendments,CLIA)和美国病理学家协会(College of American Pathologists,CAP)实验室管理相关指导文件,同时参考国家药品监督管理局(National Medical Products Administration,NMPA)、中国合格评定国家认可委员会(China National Accreditation Service for Conformity Assessment,CNAS)和美国食品药品监督管理局(U.S. Food and Drug Administration,FDA)等机构对体外诊断试剂认证原则,进行mNGS的性能确认。该文立足于北京协和医院检验科mNGS的建设经验和国内外相关研究,提出临床开展前的单中心性能确认方案。
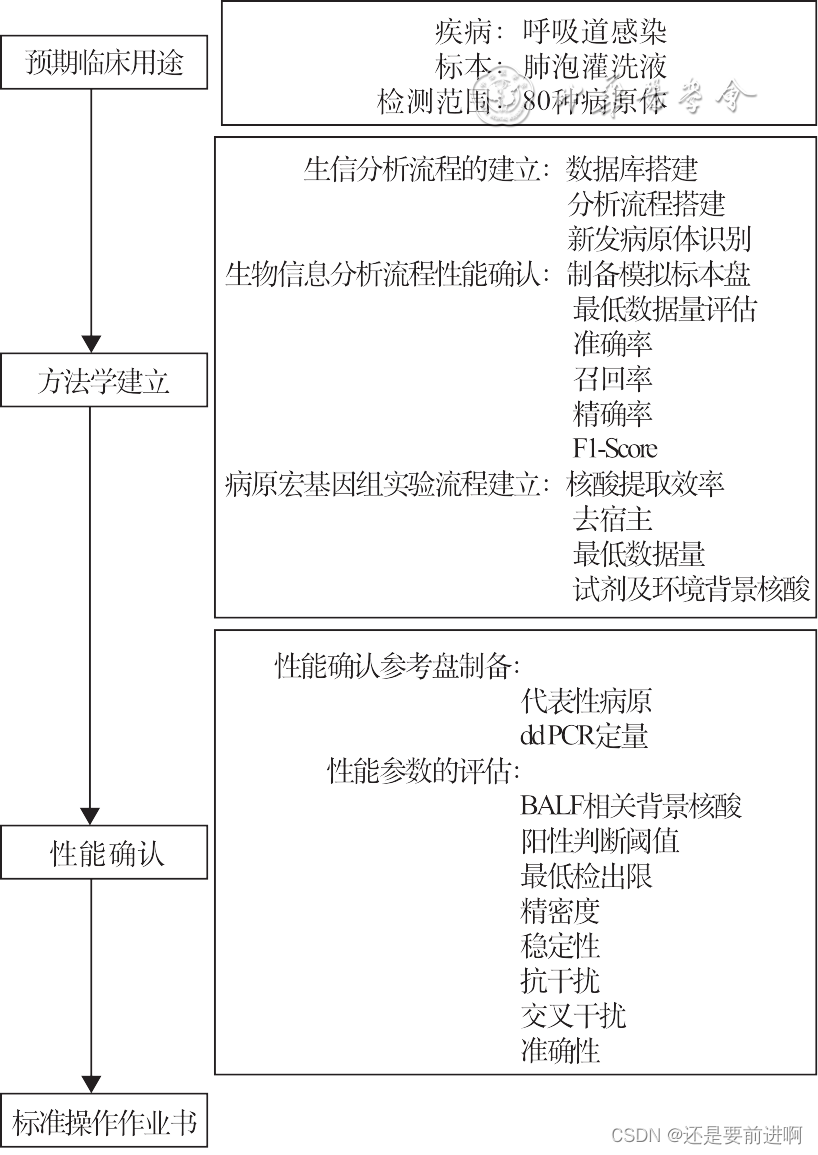
协和方案流程图(来自其它网站)
二:协和生信流程
mNGS检测涉及标本类型、检测流程和病原体众多。因此医院在mNGS方法学建立和性能确认时,建议根据临床预期用途,确定标本类型和病原体范围。
呼吸道感染是重要的感染性疾病之一,中国每年有约400万人次重症肺炎患者。国内的一项重症医学研究结果表明,超过60%的脓毒症患者由肺炎诱发。而细菌、真菌、病毒引起的肺炎多以侵染终末细支气管、肺泡等结构为主,因此肺泡灌洗液(bronchoalveolar lavage fluid,BALF)的病原学检测结果准确性更高。除此之外,BALF中定植菌和宿主细胞组成相对复杂,病原类别涵盖细菌、真菌、病毒、寄生虫和非典型病原体等。所以BALF标本可作为全面评估mNGS的代表性标本类型。
mNGS具有对不同病原体进行无偏倚检测的潜力,但评估数万种微生物的实验计划存在较大难度,有针对性地对临床重点关注的病原体进行有效性评估可行性更高。同时,建议参考传统的商品化分子生物学检测试剂的性能指标,以建立相对合理的mNGS分析性能。例如:细菌的最低检测限不高于3 000~4 000 菌落形成单位(colony forming units,CFU)/ml,病毒的最低检测限不高于1 000拷贝数/ml。
协和方案:(1)优先选择BALF标本作为mNGS性能确认的样本类型;(2)参考约翰霍普金斯大学ABX指南(https://www.hopkinsguides.com/hopkins/index/Johns_ Hopkins_ABX_Guide/Pathogens)、临床微生物学手册(第十二版)和哈里森感染病学(第七版),筛选导致肺炎的80种常见病原体;(3)细菌和病毒最低检测限建议达到1 000拷贝/ml或更低水平。
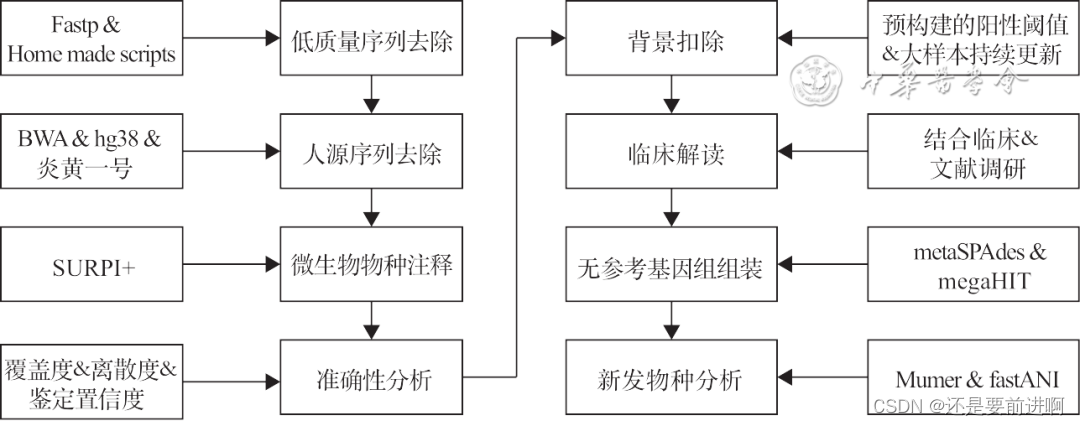
协和生信流程(来自于其它网站)。
病原宏基因分析主要涵盖以下4个模块:(1)低质量序列过滤(接头序列,测序质量低的序列,含有N的序列,低复杂度序列);(2)宿主序列过滤;(3)物种注释;(4)阳性检出判定。
mNGS生信分析流程的搭建需要计算机和生物信息学专业知识。缺少专业生物信息分析员工的实验室,建议使用开源的分析流程,如SUPRI+或经过大量临床标本验证的成熟商业分析软件,并进行性能确认。同时,在建立生物信息流程时,建议建立病原基因组的从头组装和进化分析流程。
协和方案:(1)选择SE50(MGI)或SE75(Illumina);(2)选用fastp处理接头序列和低质量序列[17];(3)使用自建的分析脚本去除含N的序列;(4)选用人类参考基因组hg38、hg19和人线粒体参考基因组作为宿主参考基因组,比对软件选用BWA并构建兼容的比对数据库;(5)选用开源的SUPRI+流程进行物种注释;(6)选用metaSPAdes和megaHIT进行从头组装,选用重叠群组装箱(contig integrator for sequence assembly,CISA)整合组装结果;(7)使用MuMer计算平均核苷酸相似度(average nucleotide identity,ANI);(8)使用热图方式展示ANI结果,根据ANI结果发掘潜在的新发病原体。
三、生物信息分析流程性能验证
随着宏基因组测序技术广泛应用于临床,科学研究者和临床专家均意识到生物信息分析的可靠性是保证结果准确的决定因素之一,加强流行病学中观察性研究报告质量(STrengthening the Reporting of OBservational studies in Epidemiology,STROBE)和CAP等学术组织都提出了对宏基因组生物信息分析流程验证的建议。内容主要包括:(1)验证近缘物种的相互交叉干扰率评估;(2)分析流程的灵敏度评估;(3)数据库的完整性和代表性评估。
协和方案:(1)根据制定的物种清单(80种),收集同属内所有微生物的代表基因组;(2)根据病原体种类不同,分别从病理系统资源整合中心(PAThosystems Resource Integration Center,PATRIC,https://www.patricbrc.org/)和临床级微生物数据库(Food and Drug Administration-database for Regulatory-Grade microbial Sequences,FDA- ARGOS,https://www.fda.gov/medicaldevices/science- and-research-medical-devices/databasereference-grade-microbial-sequen ces-fda-argos)中筛选细菌参考基因组,从全球共享禽流感数据倡议组织数据库(Global Initiative on Sharing Avian Influenza Data,GISAID,https://www.gisaid.org/)、病毒参考数据库(Reference Viral DataBase,RVDB,https://rvdb.dbi.udel.edu/)和美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI,https://www.ncbi.nlm.nih.gov/)中筛选病毒参考基因组,从真菌和卵菌信息网(Fungal and Oomycete Informatics Resources,FungiDB,https://fungidb.org/fungidb/app)和NCBI中筛选真菌参考基因组,从蠕虫寄生虫信息网(WormBase ParaSite,https://parasite.wormbase.org/index.html)和NCBI中筛选寄生虫参考基因组;微生物参考基因组筛选原则如下:①优先选择宿主为人类的基因组序列;②优先选择基因组更完整的数据,部分微生物无完整基因组时,优先选择部分组装的基因组序列;③优先选择中国地区上传的基因组序列;(4)使用全基因组模拟器(whole genome simulation,wgsim)制备评估用模拟数据;(5)设置数据库中的所有物种不同序列数梯度的模拟数据;(6)根据物种进化速率,模拟生成新发病原体的测序数据。
四、生物信息分析流程性能确认指标
本方案中,在参考计算机软件评估要求的基础上拟定mNGS生物信息分析流程性能确认的评估指标,主要包括准确率、召回率、精确率、F1-Score和最低测序数据量。
(一)准确率、召回率、精确率和F1-Score
1.生物信息分析阳性检出阈值的标准:(1)检出种特异性序列数至少1条;(2)与同属内检出序列数第一位物种间比值大于10%。
2. 肺泡微生态模拟数据的生成:对1 000份既往已进行mNGS检测的BALF标本进行重分析,统计标本中微生态的组成和丰度情况,使用宏基因组仿真数据生成软件(Critical Assessment of Metagenome Interpretation,CAMISIM)模拟微生态数据。
3. 模拟阳性标本测序数据的生成:随机生成10、100、1 000、10 000和100 000条病原体序列,并分别与微生态模拟数据整合。使用自建分析流程和数据库对每份模拟数据重复分析100次,分别计算:准确率=(TP+TN)/(TP+FN+FP+TN);召回率=TP/(TP+FN);精确率=TP/(TP+FP);F1-Score=2×准确率×召回率/(准确率+召回率);TP,真阳性;FP,假阳性;TN,真阴性;FN,假阴性。
(二)最低测序数据量
对上述1 000份BALF的背景微生物进行基因组大小的加权平均计算,获得背景微生物的平均基因组长度。根据人源序列和背景微生物的比例分布范围,估算背景微生物平均拷贝数。
已有数据表明,BALF宿主细胞中位值在105 细胞数/ml,极限高值为107 细胞数/ml。本方案中,拟定病原体最低拷贝数103 拷贝/ml,并考虑背景菌平均基因组和拷贝数。
根据Dirk Höper团队提供的数学模型(伯努利随机过程)推导出所需最低测序数据量。
五、mNGS实验流程建立
mNGS湿实验环节的方法学建立主要涉及核酸提取、建库等流程,在检测体系建立时,需进行充分评估。第一,不同的提取试剂对细菌、真菌、病毒的核酸提取效率存在差异。同时,提取流程中是否有破壁过程以及破壁的条件也会影响核酸提取效率。第二,根据已发表的专家共识,当临床标本中宿主细胞浓度高时,为提高低载量病原体的检出率,建议添加去宿主过程。第三,mNGS应用于科学研究时多采用单份标本最低20M的测序数据量,但临床应用时需要多少测序数据量存在较大的争议。建议在建立方法学时,进行最低测序数据量的评估。第四,背景微生物的核酸片段是干扰mNGS结果的重要因素。建议在建立方法学时,根据实际情况建立背景核酸数据模型,协和方案如下。
(一)核酸提取效率评估
使用固定CFU的代表性菌株,使用Qiagen的提取试剂盒提取核酸,并使用微滴式数字聚合酶链反应(droplet digital polymerase chain reaction,ddPCR)进行定量,每一菌株重复3次以计算均值;以阴性BALF剩余标本为基质,加入固定CFU的代表性菌株制备模拟标本,以实验室拟使用的提取流程进行核酸提取,使用ddPCR进行定量,并重复3次以计算均值;计算两者间比值,定义为提取流程对代表性菌株的提取效率。
(二)去宿主病原体损耗评估
去宿主病原体损耗评估参考盘制备:拟投入的病原体为鲍曼不动杆菌、流感嗜血杆菌、卡他莫拉杆菌、金黄色葡萄球菌、铜绿假单胞菌、肺炎克雷伯菌、肺炎链球菌、白色念珠菌、烟曲霉、表皮葡萄球菌、巨细胞病毒(Cytomegalovirus,CMV)、EB病毒(Epstein-Barr virus,EBV)和腺病毒,每种病原体浓度拟设定为1 000 拷贝/ml,每份标本中宿主细胞浓度拟控制在105 细胞数/ml。
随机选择6份模拟标本平均分为2组,一组使用去宿主试剂进行处理,另一组不进行去宿主处理,获得核酸后同时进行建库测序和ddPCR。
统计分析2组不同处理标本目标病原体每百万序列数(reads per million,RPM)和定量结果的差异。
统计分析2组标本宿主基因组比率和内参基因定量结果的差异。
(三)最低测序数据量评估
1.DNA流程最低测序数据量参考盘制备:拟投入病原体同“五(二)”中使用的病原体。分别使用宿主细胞浓度为105 细胞数/ml和107 细胞数/ml的肺泡灌洗液剩余标本作为稀释基质,建议每种病原体浓度1 000 拷贝/ml,不同宿主浓度梯度的标本建议重复检测不少于5次。
2.RNA流程最低测序数据量参考盘制备:拟投入病原体甲型流感病毒、鼻病毒、呼吸道合胞病毒和冠状病毒,每种病原体浓度为1 000 拷贝/ml(参考ddPCR结果)。宿主细胞浓度同DNA流程最低测序数据量参考盘。不同宿主浓度梯度的模拟标本建议重复检测不少于5次。
3.最低测序数据量评估:每份标本的测序数据量为400 兆(megabyte,M)reads,随机抽取12.5、25、50和100 M各20次分别进行生信分析流程进行分析,通过probit回归分析合理的最低测序数据量。
(四)建立试剂及环境背景核酸数据库
1.试剂工程菌背景核酸的建立:(1)酒精清洁双手,建议准备好2 ml EP管12个,在试剂准备间超净工作台内,每个EP管中分装1 ml生理盐水并编号;(2)将标本随机平分为2组,分别使用去宿主流程和不去宿主流程进行测序;(3)测序后,分析去宿主流程和不去宿主流程的微生物检出情况和相对丰度。
2.实验室空间和设备表面背景核酸的建立:(1)酒精清洁双手,按照实验室实际情况准备2 ml EP管若干,在核酸提取区生物安全柜中分装1 ml 生理盐水至每个EP管中并编号。(2)使用无菌拭子分别对每个实验区的台面和设备(实验台面、生物安全柜、离心机孔位及表面、均质仪孔位及表面、PCR扩增仪孔位及表面、涡旋震荡仪表面、恒温金属浴孔位及表面、测序仪表面、操作人员手部、口罩外层等)进行擦拭采集环境标本,将拭子装入EP管中进行振荡混匀。建议同一位置连续3 d进行标本采集。为便于数据统计,建议每批次实验设置1份未采集环境标本的拭子作为空白对照。(3)测序后,分析各位置的微生物组成和相对丰度,建立实验室操作空间和设备背景核酸数据模型。
(五)质量控制点和风险控制点设置
建议测定提取核酸的浓度和纯度,DNA/RNA的A260/A280比值不低于1.8/2.0。文库构建完成时再次测定核酸浓度和纯度,当文库浓度低于0.1 ng/μl时,建议重新实验。同时,建议测序数据量和测序质量值作为质量控制点之一,当测序数据量低于最低测序量或Q30低于80%时,建议重复检测。同时,本方案建议在标本中加入经过ddPCR定量摩尔浓度的大肠杆菌噬菌体(DNA流程使用T7噬菌体,RNA流程使用M2噬菌体)作为内标;建议每轮实验同时检测3倍最低检出限(limit of detection,LoD)浓度的弱阳性质控、10倍LoD浓度的阳性质控和阴性质控。
六、mNGS全流程性能确认参考盘
目前,对于病原mNGS性能确认参考盘的制备缺少统一和规范的标准。国外针对呼吸道标本和脑脊液标本的参考盘制备方法是在阴性的临床标本中加入梯度稀释的临床阳性标本、病原体培养物或病原体核酸。该方法保证了参考盘接近临床标本的理化性质和微生态特征,但无法固定宿主细胞率,且临床标本异质性高,标本间微生态存在差异。不同的参考盘配置方法各有利弊,本方案中建议使用宿主细胞混合病原体或临床阳性标本的策略。
(一)DNA流程性能确认参考盘制备
使用mNGS检测为阴性的肺泡灌洗液剩余标本和生理盐水作为稀释基质,建议宿主细胞浓度设定为105 细胞数/ml;分别投入“五(二)”中使用的病原体,建议病原体浓度梯度设定为0(空白)、160、800、4 000、20 000和100 000拷贝/ml。
(二)RNA流程性能确认参考盘制备
使用mNGS检测为阴性的肺泡灌洗液剩余标本和生理盐水作为稀释基质,按照“六(一)”浓度梯度,分别投入“五(三)2”中使用的病原体。
七、mNGS全流程性能确认
对于mNGS检测,标本采集和转运过程、宿主细胞浓度等可影响背景核酸、阳性判断阈值和LoD等性能。建议针对一般情况,初步建立检测体系的阳性判断阈值、LoD、精密度、抗干扰、交叉反应情况,并对标本的稳定性和临床实际应用时检测体系的准确性进行初步探讨。
(一)BALF相关背景核酸、阳性判断阈值和LoD
使用实验室建立的实验流程对制备的参考盘进行检测,分别统计不同病原体的RPM,结合试剂及环境背景核酸模型,分别绘制相应的受试者工作特征曲线(receiver operating characteristic curve,ROC曲线)以确立相对合理的阳性判断阈值。使用probit回归计算不同病原体的LoD。
对于暂时无法或难以获取的其他病原体,建议通过统计阴性BALF标本、试剂及环境背景核酸中各病原体的RPM,以均值加3倍标准差作为阳性判断阈值。
(二)精密度确认
1.使用同批次试剂:对弱阳性标本(3倍LoD浓度)和阳性标本(10倍LoD浓度)分别进行20次重复检测并统计RPM,建议每天重复5次检测,连续4 d完成评估。
2.使用不同批次试剂:对弱阳性标本和阳性标本分别进行20次重复检测并统计RPM,建议每天重复5次检测,连续4 d完成评估。
(三)抗干扰确认
调整弱阳性标本的宿主细胞浓度至105、106、107细胞数/ml 3个浓度梯度,分别进行测序。以弱阳性质控无法检出时内参的RPM值为抗宿主干扰阈值。当进行临床报告解读时,若内参的RPM超出阈值,需警惕假阴性。
(四)交叉干扰确认
选取若干种近源物种,例如金黄色葡萄球菌和表皮葡萄球菌、白色念珠菌和热带念珠菌,将两对微生物分别按照1∶1、9∶1、1∶9的浓度比例进行混合后测序;统计分析近源物种的检出情况和比例与预期是否一致。
(五)稳定性确认
将弱阳性标本和阳性标本在4 ℃和‒20 ℃分别保存1、4、7 d之后进行检测,统计RPM;一般认为标本在‒80 ℃可在较长时间内稳定保存。本方案建议评估‒80 ℃保存的标本反复冻融时对结果的影响。分别评估在‒80 ℃保存的标本冻融1和2次后检测结果的RPM变化情况。
(六)临床准确性评估
建议收集既往mNGS检测结果为阴性的BALF剩余标本50份,阳性剩余标本20份,分别进行DNA流程和RNA流程检测。对于阳性标本,建议与既往培养结果或使用PCR复核,统计总符合率、阳性符合率和阴性符合率。
八、mNGS全流程标准操作作业书(standard operating procedure,SOP)建立。
完成性能确认后,建议根据建立的实验流程,编写可读性强的SOP。建议分别制定DNA流程和RNA流程的SOP,涵盖实验操作具体步骤及注意事项、质量控制点及操作处理指示、实验记录的规范和注意事项、数据分析及解读流程、数据备份及安全管理指示和管理员权限等重要的具体细节,保证全流程的操作规范、可重复和可溯源。
近年来,mNGS技术对临床感染性疾病的诊断,尤其是在新发突发病原体比如新型冠状病毒的鉴定中发挥了重要作用。但目前病原mNGS的性能确认、质量控制和规范化的管理,学术界仍然有较大空白。为保证临床检测结果的可靠性,本方案中提出的性能确认流程相对复杂。尽管性能确认不能解决mNGS技术中存在的诸多问题,但能够为mNGS的工作人员提供诸如核酸提取效率、去宿主效率、数据库、生物信息学分析流程、背景核酸和阳性判断阈值等相关参数信息,同时能够明确全流程的精密度、LoD、抗干扰、交叉反应和稳定性等相关性能,这些信息对于mNGS报告解读具有重要的指导价值。本方案中仍存在不足和错误之处,希望同行批评指正,继续完善性能确认方案,为mNGS的临床规范化应用贡献更多的智慧和力量。